|
|||||


 LAGOTRAN® 25 mg / 50 mg / 100 mg / 200 mg LAGOTRAN® 25 mg / 50 mg / 100 mg / 200 mg
|
BETA | |
| Lamotrigina | ||
|
Anticonvulsivantes Antiepilépticos [Sistema Nervioso Central] |
||
|
Venta Bajo Receta Archivada Comprimidos Dispersables Industria Argentina |
||
|
||
|
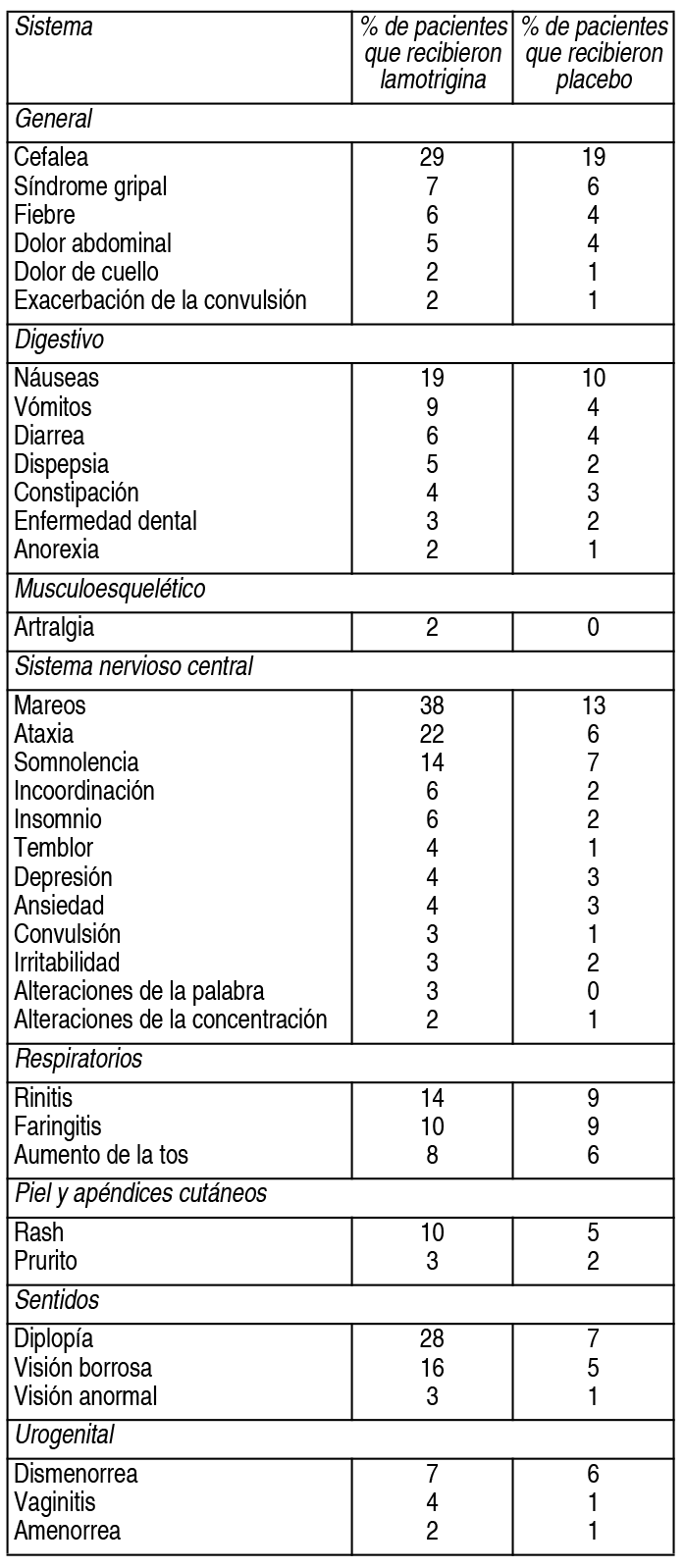
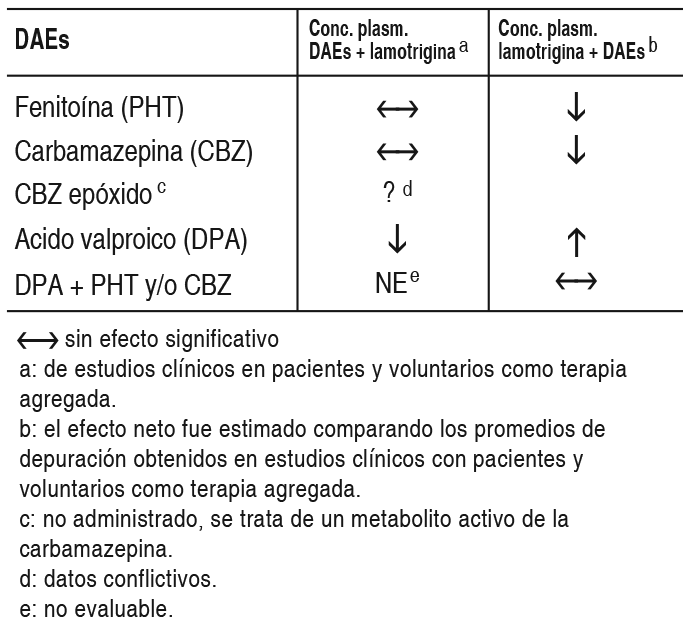
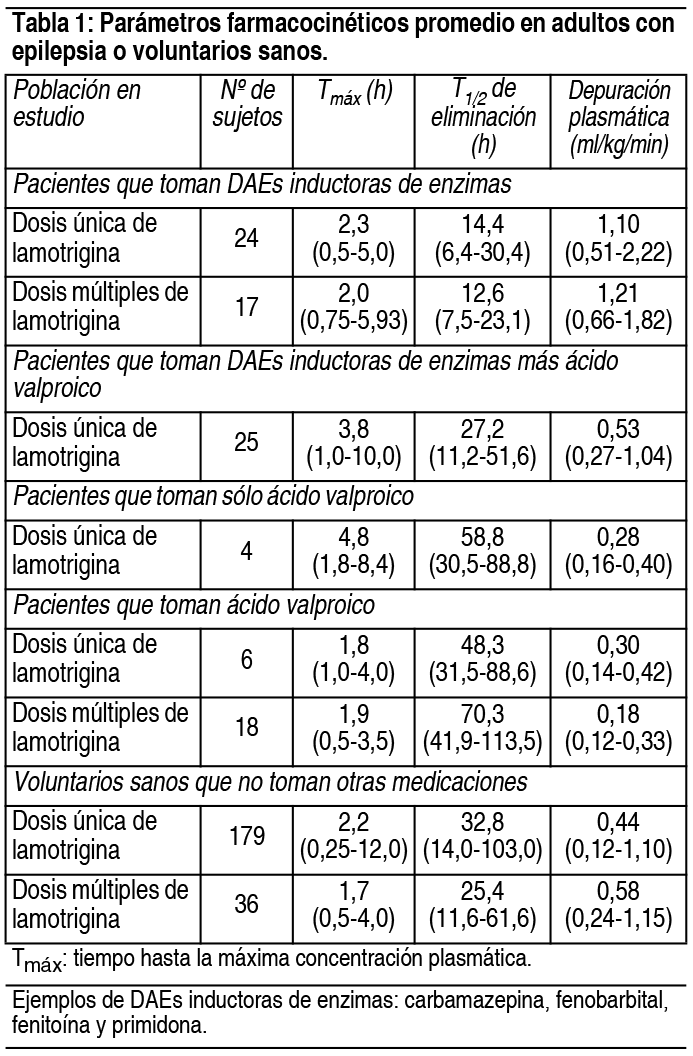
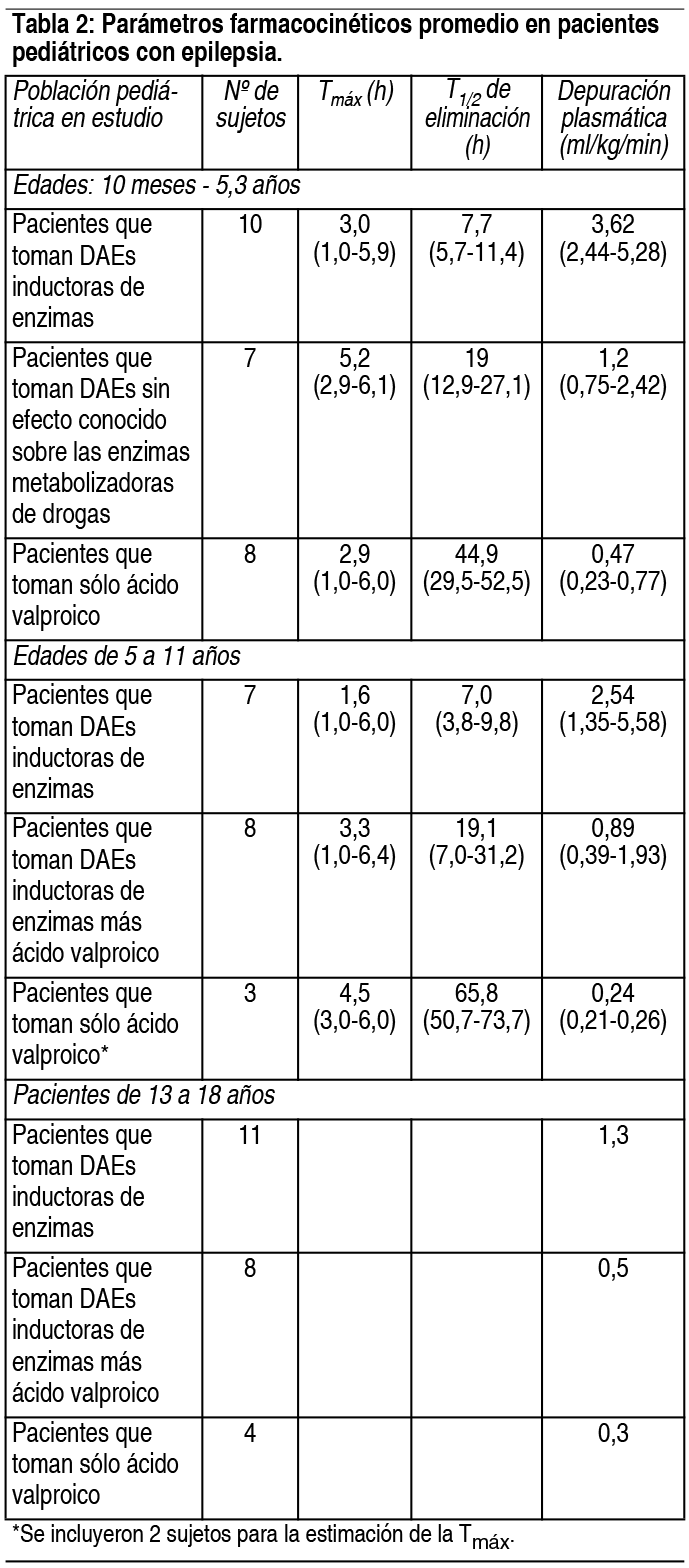
COMPOSICION: Lagotran® 25 mg: Cada comprimido dispersable contiene: Lamotrigina 25 mg. Excipientes: Lactosa, Crospovidona, Laurilsulfato de Sodio, Glicolato Sódico de Almidón, Sacarina Sódica, Esencia de Frutilla, Ludipress, Povidona y Estearato de Magnesio c.s. Lagotran® 50 mg: Cada comprimido dispersable contiene: Lamotrigina 50 mg. Excipientes: Lactosa, Crospovidona, Laurilsulfato de Sodio, Glicolato Sódico de Almidón, Sacarina Sódica, Esencia de Frutilla, Ludipress, Povidona y Estearato de Magnesio c.s. Lagotran® 100 mg: Cada comprimido dispersable contiene: Lamotrigina 100 mg. Excipientes: Lactosa, Crospovidona, Laurilsulfato de Sodio, Glicolato Sódico de Almidón, Sacarina Sódica, Esencia de Frutilla, Ludipress, Povidona y Estearato de Magnesio c.s. Lagotran® 200 mg: Cada comprimido dispersable contiene: Lamotrigina 200 mg. Excipientes: Lactosa, Crospovidona, Laurilsulfato de Sodio, Glicolato Sódico de Almidón, Sacarina Sódica, Esencia de Frutilla, Ludipress, Povidona y Estearato de Magnesio c.s. ACCION: Antiepiléptico. INDICACIONES: Terapia coadyuvante: La lamotrigina está indicada como tratamiento coadyuvante en adultos con crisis parciales y en las crisis generalizadas del síndrome de Lennox-Gastaut tanto en pacientes pediátricos como en adultos. Monoterapia: La lamotrigina está indicada para conversión a la monoterapia en pacientes adultos con crisis parciales que se encuentran recibiendo tratamiento con una única droga antiepiléptica (DAE) inductora de enzimas. La seguridad y eficacia de la lamotrigina no han sido establecidas: 1) como monoterapia inicial, 2) para la conversión a monoterapia de tratamientos con fármacos antiepilépticos no inductores de enzimas (por ejemplo valproato) o 3) para conversión simultánea a monoterapia de 2 o más drogas antiepilépticas en forma concomitante. La seguridad y eficacia en pacientes menores de 16 años que no padezcan síndrome de Lennox-Gastaut no han sido establecidas. DOSIS: Terapia coadyuvante: La lamotrigina está indicada como tratamiento coadyuvante en adultos con crisis parciales y en las crisis generalizadas del síndrome de Lennox-Gastaut tanto en pacientes pediátricos como en adultos. Monoterapia: La lamotrigina está indicada para conversión a la monoterapia en pacientes adultos con crisis parciales que se encuentran recibiendo tratamiento con una única DAE inductora de enzimas (por ejemplo: carbamazepina, fenitoína, fenobarbital, etc). La seguridad y eficacia de la lamotrigina no han sido establecidas: 1) como monoterapia inicial, 2) para la conversión a monoterapia de tratamientos con fármacos antiepilépticos no inductores de enzimas (por ejemplo valproato) ó 3) para conversión simultánea a monoterapia de 2 o más drogas antiepilépticas en forma concomitante. La seguridad y eficacia en pacientes menores de 16 años que no padezcan síndrome de Lennox-Gastaut no han sido establecidas. Consideraciones generales para establecer la dosis: el riesgo de aparición de rash está aumentado cuando la dosis de inicio de la lamotrigina es mayor a la recomendada y/o cuando se excede la tasa de incremento de dosis sugerida. Se ha sugerido, pero aún resta ser probado, que el riesgo de padecer rash potencialmente fatal parece aumentar en las siguientes situaciones: 1) pacientes tratados en forma simultánea con ácido valproico y lamotrigina, 2) cuando se excede la dosis inicial recomendada y 3) cuando se excede el incremento de dosis sugerido. Por lo tanto, es de suma importancia respetar estrechamente las recomendaciones de dosis indicadas. Dosis en terapia coadyuvante: En este apartado se describen las recomendaciones de dosis específicas para adultos y niños mayores de 12 años por un lado, y para pacientes con edades comprendidas entre 2 y 12 años por el otro. Además de considerar el grupo etario correspondiente, cada recomendación tiene en cuenta si el paciente se encuentra recibiendo o no valproato. Además, se sugieren los esquemas posológicos para aquellos pacientes que se encuentran recibiendo en forma concomitante DAEs que no han sido sistemáticamente evaluadas en combinación con la lamotrigina. En los esquemas de dosificación que se describen a continuación, las DAEs inductoras de enzimas incluyen a fenitoína, carbamazepina, fenobarbital y primidona. Niños de 2 a 12 años: El esquema de dosificación recomendado de lamotrigina en pacientes que están recibiendo valproato se muestra en la siguiente tabla: Ver Tabla El régimen de dosis recomendado de lamotrigina en pacientes que están recibiendo DAEs inductoras de enzimas se esquematiza en la tabla 4. Ver Tabla Pacientes mayores de 12 años: El esquema de dosificación recomendado de lamotrigina en pacientes que están recibiendo valproato se muestra en la siguiente tabla. Ver Tabla El régimen de dosis recomendado de lamotrigina en pacientes que están recibiendo DAEs inductoras de enzimas se esquematiza en la tabla 6. Ver Tabla Pasaje de un régimen con una única DAE inductora de enzimas a monoterapia con lamotrigina en pacientes de 16 años o más: El objetivo del régimen de transición es realizar el pasaje a monoterapia con lamotrigina en condiciones que aseguren el adecuado control de las crisis mitigando el riesgo de aparición de rash asociado con el rápido incremento de la dosis del fármaco. El pasaje a monoterapia se realiza en dos etapas. En la primera, se van realizando los incrementos de lamotrigina hasta alcanzar la dosis deseada mientras se mantiene la DAE inductora de enzimas en una dosis fija. En una segunda etapa, la DAE inductora de enzimas se va suspendiendo en forma gradual en un período de 4 semanas. La dosis recomendada de mantenimiento de lamotrigina es de 500 mg/día, administrada en 2 tomas. Para alcanzar la dosis de 500 mg/día, la lamotrigina debe agregarse a un régimen de tratamiento con una DAE inductora de enzimas según el esquema mencionado en la tabla 6. El régimen de suspensión de la DAE inductora de enzimas concomitante se basa en la experiencia obtenida en el ensayo clínico controlado de monoterapia. En ese ensayo, la DAE inductora de enzimas se disminuyó a razón de 20% de la dosis por semana en un período de alrededor de 4 semanas. Debido al incremento del riesgo de rash, no deben excederse la dosis inicial y los posteriores aumentos de dosis de lamotrigina recomendados. Dosis usual de mantenimiento: las dosis usuales de mantenimiento mencionadas en las tablas anteriores, derivan de regímenes de dosis empleados en ensayos controlados con placebo de terapia coadyuvante, en los cuales se estableció la eficacia de la lamotrigina. En pacientes que recibieron regímenes con varios fármacos entre los cuales se emplearon DAEs inductoras de enzimas sin valproato, se utilizaron dosis de mantenimiento de lamotrogina de hasta 700 mg/día como tratamiento coadyuvante. En pacientes que recibieron solamente valproato, se utilizaron dosis de hasta 200 mg/día como terapia de mantenimiento en tratamientos coadyuvantes con lamotrigina. La ventaja del empleo de dosis superiores a las recomendadas en las tablas que figuran más arriba, no ha sido establecida en ensayos controlados. Lamotrigina como tratamiento coadyuvante en pacientes que se encontraban recibiendo otras DAEs diferentes a DAEs inductoras de enzimas y a valproato: El efecto de otras DAEs diferentes a DAEs inductoras de enzimas y a valproato sobre el metabolismo de la lamotrigina no puede predecirse. De todos modos no se pueden administrar recomendaciones específicas de dosis en tales situaciones. Es prudente respetar las recomendaciones de inicio y de aumento de dosis para pacientes que se encuentren recibiendo valproato. La dosis de mantenimiento estará comprendida entre el valor de la dosis de mantenimiento en los pacientes que se encuentren recibiendo valproato y el valor de la dosis de mantenimiento en pacientes que se encuentren recibiendo DAEs inductoras de enzimas sin valproato. Pacientes con deterioro hepático: la experiencia en pacientes con deterioro hepático es limitada. Las siguientes recomendaciones generales están hechas en base a un estudio clínico farmacológico realizado en pacientes con disfunción hepática moderada a severa. La dosis de inicio, los aumentos posteriores de dosis y la dosis de mantenimiento deberán reducirse en aproximadamente un 50% en pacientes con deterioro hepático moderado (índice de Child-Pugh B) y en un 75% en pacientes con deterioro hepático severo (índice de Child-Pugh C). Los aumentos de dosis y la dosis de mantenimiento deberán ajustarse de acuerdo a la respuesta clínica. Pacientes con deterioro funcional renal: las dosis de inicio en el tratamiento con lamotrigina deben respetar los esquemas indicados anteriormente. Una dosis de mantenimiento reducida puede ser efectiva para pacientes con deterioro funcional renal significativo. Algunos pacientes con severo deterioro renal han sido evaluados durante el tratamiento en forma crónica con lamotrigina. Debido a que no existe suficiente experiencia sobre el empleo de lamotrigina en esta población, este fármaco deberá emplearse con precaución en estos pacientes. Estrategia de discontinuación: en aquellos pacientes que estén recibiendo lamotrigina en combinación con otras DAEs, el régimen antiepiléptico deberá reevaluarse para la totalidad de las drogas si aparecieran cambios en el control de las crisis o empeora- miento de los eventos adversos. Si se tomara la decisión de discontinuar la terapia con lamotrigina, deberá realizarse la disminución de la misma en un período de 2 semanas (aproximadamente 50% de la dosis por semana) a menos que por cuestiones de seguridad sea necesario un más rápido retiro del fármaco. La suspensión de una DAE inductora de enzimas puede prolongar la vida media de la lamotrigina. La suspensión de la administración de valproato puede acortar la vida media de la lamotrigina. Niveles plasmáticos: no se ha establecido un rango de concentraciones plasmáticas terapéuticas de lamotrigina. La dosis de lamotrigina deberá ajustarse en función de la respuesta terapéutica. Administración de los comprimidos dispersables de lamotrigina: los comprimidos de lamotrigina pueden ser ingeridos enteros, masticarse o dispersarse en agua o jugo de frutas diluido. Si los comprimidos se mastican, se recomienda ingerirlos con una pequeña cantidad de agua o jugo de frutas diluido para facilitar su deglución. Para disolver los comprimidos de lamotrigina, agregue a los mismos una pequeña cantidad de líquido (una cucharada de té o la cantidad suficiente como para cubrir la medicación). Aproximadamente 1 minuto después, cuando los comprimidos estén totalmente deshechos, revuelva la solución y consuma todo inmediatamente. No deben realizarse intentos de administrar cantidades parciales de los comprimidos dispersables. CONTRAINDICACIONES: Lagotran® está contraindicado en pacientes con hipersensibilidad conocida a la lamotrigina o a cualquiera de los componentes del producto. ADVERTENCIAS: Si bien durante el tratamiento con lamotrigina puede presentarse rash de naturaleza leve, debido a que no es posible predecir la gravedad del mismo, el fármaco deberá suspenderse ante el primer signo de rash, a menos que la aparición de éste, claramente no esté relacionada con la droga. La suspensión del tratamiento puede no prevenir las secuelas que aparecieran a consecuencia del mismo y/o las complicaciones que podrían poner en compromiso la vida. Reacciones cutáneas severas: Se han informado reacciones cutáneas adversas, las cuales generalmente se presentan dentro de las primeras 8 semanas de tratamiento. La mayoría de las reacciones cutáneas son leves y autolimitadas; no obstante, pueden ocurrir raramente reacciones cutáneas severas y potencialmente fatales, incluyendo síndrome de Stevens-Johnson y necrólisis epidérmica tóxica. La incidencia aproximada de las reacciones cutáneas severas en adultos y niños mayores de 12 años es de 1 en 1.000. Sin embargo, no todos los casos pueden ser atribuidos a la droga: el 5% de pacientes expuestos a placebo también desarrollan rash cutáneo. Todos los pacientes (adultos y niños) que desarrollen un rash cutáneo deberían ser rápidamente evaluados y la lamotrigina suspendida, a menos que el rash no esté claramente relacionado con la droga. El inicio benigno de rash no indica una evolución totalmente benigna, sin embargo, un número significativo de pacientes que han desarrollado rash con lamotrigina han continuado el tratamiento sin complicaciones. Antes del comienzo del tratamiento los pacientes deben ser advertidos sobre: 1) el rash puede ocurrir; 2) puede ser el anuncio de un evento médico serio; 3) que de ocurrir debe ser informado inmediatamente a su médico. En 3.400 sujetos que participaron en ensayos clínicos, la incidencia de rash que requirió internación fue del 0.3%. En estos individuos no ocurrieron episodios fatales, pero en informes obtenidos durante la comercialización, el rash ha sido asociado con casos fatales. Entre los rash que requirieron internación, hubo casos de síndrome de Stevens-Johnson, necrólisis epidérmica tóxica, angioedema y rash asociado con fiebre, linfoadenopatía, edema facial, anormalidades hematológicas y hepáticas. Existe evidencia de que la inclusión de valproato en terapia combinada, incrementa el riesgo de rash severo y amenazador para la vida. Más específicamente, de 584 pacientes tratados con lamotrigina junto con valproato en ensayos clínicos, 6 (1%) fueron hospitalizados por rash; por otra parte, 4 (0.16%) de 2.398 pacientes y voluntarios tratados con lamotrigina sin valproato asociado, requirieron internación. También ocurrieron otros ejemplos de rash severo y potencialmente amenazador para la vida que no requirieron internación en la fase de precomercialización. Entre ellos, un caso fue reportado como símil Stevens-Johnson. Rash en niños: la incidencia de rash severo asociado con hospitalización y discontinuación del tratamiento con lamotrigina detectada en estudios prospectivos realizados en pacientes pediátricos fue del 1,1%. No se produjeron muertes ni secuelas permanentes en estos pacientes. Raramente se detectaron casos de necrólisis epidérmica tóxica. Si bien el riesgo parece ser similar tanto en pacientes pediátricos que reciben concomitantemente valproato como en aquellos que no lo reciben, algunos reportes obtenidos luego de la comercialización del producto sugieren que el porcentaje de rash severo fue mayor en pacientes que emplearon valproato en forma concomitante. Se debe remarcar que la lamotrigina sólo está indicada en aquellos pacientes de menos de 16 años que tienen convulsiones asociadas con el síndrome de Lennox-Gastaut (ver Indicaciones). Retiro de lamotrigina debido a rash: no hay indicadores de severidad certeros, el retiro de lamotrigina debido a rash es una decisión médica. La discontinuación más común fue en pacientes bajo regímenes de drogas que incluían valproato pero aún más frecuente cuando la lamotrigina fue coadministrada sólo con valproato. Como el rash ocurre frecuentemente asociado con otros signos y síntomas es imposible discernir en qué proporción el mismo fue la causa principal para el retiro de la droga. La proporción global de discontinuación debido a rash en pacientes que participaron en estudios clínicos (n = 3.501) fue 3.8%. Para determinar la importancia del rash, debería considerarse que un número significativo de pacientes con erupción cutánea continuaron con el tratamiento sin presentar ningún evento. Reacciones de hipersensibilidad/falla multiorgánica: Se han descrito reacciones cutáneas como parte de un síndrome de hipersensibilidad asociado con un patrón variable de síntomas sistémicos, incluyendo fiebre, linfoadenopatía, edema facial y anormalidades hematológicas y hepáticas. El síndrome muestra un amplio espectro de severidad y puede, raramente, conducir a una coagulación intravascular diseminada y a una falla multiorgánica. Es importante notar que las primeras manifestaciones de hipersensibilidad (por ej.: fiebre, linfoadenopatía, pueden estar presentes aún cuando el rash no es evidente). Los pacientes deberían ser advertidos acerca de la necesidad de una consulta médica inmediata ante la aparición de los mencionados signos y síntomas. En presencia de ellos, el paciente debería ser evaluado inmediatamente y la medicación discontinuada si un diagnóstico alternativo no puede ser establecido. Ha sido reportado un caso de insuficiencia hepática fulminante en una paciente de 23 años recibiendo concomitantemente ácido valproico y carbamazepina. La misma tuvo cefalea, fiebre y rash maculopapular luego de 3 semanas de haber iniciado el tratamiento con lamotrigina. Dentro de los 3 días posteriores, independientemente de una subsecuente mejora clínica aparente, presentó coma hepático. Dos meses después y de forma inesperada, tuvo un desenlace fatal por causa de una embolia pulmonar. Casos fatales asociados con falla multiorgánica y diferentes grados de insuficiencia hepática han sido informados en 5 pacientes de los 7.000 que recibieron lamotrigina durante la fase de precomercialización. Esos casos ocurrieron conjuntamente con otros eventos médicos serios (por ej.: status epilepticus, sepsis fulminante) haciendo imposible identificar la causa primaria. Además, sin ningún evento precipitante evidente, una mujer de 45 años tratada con carbamazepina y clonazepam presentó rabdomiolisis, insuficiencia renal, rash, ataxia y elevación de AST, 14 días después de haber sido adicionada lamotrigina a su régimen antiepiléptico. Subsecuentemente se recuperó con terapia de soporte luego de haberse discontinuado la lamotrigina. También han ocurrido raramente, durante la experiencia clínica con lamotrigina como terapia adjunta a otra droga, enfermedades rápidamente progresivas con estado epiléptico, rabdomiolisis, disfunción multiorgánica y coagulación intravascular diseminada, que han llevado a la muerte. La contribución de la lamotrigina en estos cuadros resta por ser establecida. Convulsiones por retiro de droga: Como regla, las drogas antiepilépticas no deben ser suspendidas abruptamente por la posibilidad de que aumenten las convulsiones. A menos que exista un problema que requiera su rápido retiro, la dosis de lamotrigina debe ser disminuida durante un período de 2 semanas. Eventos hematológicos: La lamotrigina es un inhibidor débil de la dihidrofolato reductasa, por lo cual existe la posibilidad de interferencia con el metabolismo del folato durante la terapia prolongada. No obstante, no han ocurrido alteraciones de la concentración de hemoglobina, el volumen corpuscular medio o las concentraciones de folato a nivel sérico o eritrocitario en tratamientos de hasta 1 año o de las concentraciones de folato eritrocitario en tratamientos de hasta 5 años. Se ha publicado un caso de aplasia pura de eritrocitos en una persona de sexo masculino de 32 años de edad con antecedentes de talasemia ß. La aspiración de médula ósea reveló un marcado descenso de la eritropoyesis (con granulocitopoyesis y trombopoyesis normal), la cual revirtió luego de la discontinuación del tratamiento y la transfusión de glóbulos rojos. EFECTOS: Las experiencias adversas más comúnmente observadas asociadas con el uso de lamotrigina en combinación con otras DAEs, que no fueron vistas con una frecuencia equivalente en los pacientes tratados con placebo, fueron mareos, ataxia, somnolencia, cefalea, diplopía, visión borrosa, náuseas, vómitos y rash cutáneo. Los mareos, la diplopía, la ataxia y la visión borrosa ocurrieron más en pacientes que recibieron carbamazepina con lamotrigina, que en pacientes que recibieron otras DAEs con lamotrigina. Datos clínicos sugieren una mayor incidencia de rash cutáneo, incluyendo rash severo en pacientes que reciben ácido valproico concomitantemente que en pacientes que no lo reciben. Alrededor del 11% de 3.378 pacientes que recibieron lamotrigina como terapia coadyuvante en ensayos de premarketing discontinuaron el tratamiento debido a la aparición de algún evento adverso. Los eventos adversos más comúnmente asociados con la discontinuación del tratamiento fueron rash (3%), mareos (2.8%) y cefalea (2.5%). En un estudio de respuesta de dosis la suspensión de lamotrigina por mareos, ataxia, diplopía, visión borrosa, náuseas y vómitos estuvo relacionada con la dosis. Monoterapia en adultos: Los eventos adversos más comúnmente observados (≥ 5%) relacionados al uso de la lamotrigina en monoterapia en adultos y no observados a una tasa equivalente en el grupo control fueron: Generales: infección, dolor y dolor de pecho. Metabolismo: descenso de peso. Aparato digestivo: vómitos, dispepsia y náuseas. Sistema nervioso: coordinación anormal, mareos, ansiedad e insomnio. Aparato respiratorio: rinitis. Aparato genitourinario: dismenorrea. Los eventos adversos más comúnmente observados (≥ 5%) relacionados al uso de lamotrigina durante la conversión a monoterapia fueron: Generales: astenia y prurito. Sistema nervioso: mareos, cefalea, coordinación alterada, rash, somnolencia, diplopía, ataxia, temblor, visión borrosa, insomnio y nistagmo. Aparato digestivo: diarrea, náuseas y vómitos. Sistema hemolinfático: linfoadenopatía. Aparato respiratorio: sinusitis. Otros eventos adversos observados en al menos un 2% de los pacientes tratados con lamotrigina y numéricamente más frecuentes que en el grupo de valproato fueron: Generales: dolor, infección, dolor torácico, diaforesis y fiebre. Aparato digestivo: dispepsia, anorexia, sequedad bucal, hemorragia rectal, úlcera péptica, disminución de peso y edema periférico. Sistema nervioso: depresión, ansiedad, amnesia, hipoestesia, aumento de la libido, disminución de reflejos, irritabilidad e ideación suicida. Aparato respiratorio: rinitis, epistaxis, bronquitis y disnea. Piel y apéndices: dermatitis de contacto y piel seca. Sistema genitourinario: dismenorrea. Aproximadamente un 10% de los pacientes que participaron en los ensayos de lamotrigina como monoterapia abandonaron el tratamiento, siendo los eventos adversos más comúnmente asociados con la discontinuación: rash (4,5%), cefalea (3.1%) y astenia (2.4%). Los eventos adversos observados en al menos un 2% de los pacientes tratados con lamotrigina como tratamiento coadyuvante, y numéricamente más frecuentes que en el grupo placebo en pacientes adultos y pediátricos con síndrome de Lennox-Gastaut fueron: Generales: infección, lesión accidental, síndrome gripal y astenia. Aparato digestivo: dolor abdominal, hemorragia, vómitos, constipación, anorexia, diarrea y náuseas. Sistema nervioso: ataxia, convulsiones y temblor. Aparato respiratorio: faringitis, bronquitis y neumonía. Piel y apéndices: rash y eczema. Sistema genitourinario: infección urinaria, balanitis y trastornos penianos. Incidencia en ensayos clínicos controlados en adultos de lamotrigina como tratamiento coadyuvante: La siguiente tabla enumera los eventos adversos aparecidos en al menos el 2% de los pacientes con epilepsia tratados con lamotrigina agregada a un régimen con otras DAEs. Ver Tabla En un ensayo clínico doble ciego de terapia coadyuvante, los rash cutáneos se presentaron en hasta el 10% de los pacientes que tomaban lamotrigina y en el 5% de los pacientes que tomaron placebo. La aparición de rash cutáneo motivó la suspensión del tratamiento con lamotrigina en el 2% de los pacientes. El rash, usualmente de apariencia maculopapular generalmente aparece dentro de las 8 semanas de comenzado el tratamiento y desaparece con la suspensión de la lamotrigina. Raramente se han reportado rash cutáneos serios, potencialmente fatales, incluyendo el síndrome de Stevens-Johnson y la necrólisis epidérmica tóxica (síndrome de Lyell). Adicionalmente, el riesgo de aparición de rash está asociado con: Dosis de inicio y aumento de dosis de la lamotrigina que exceden el esquema recomendado. Uso concomitante de valproato que aumenta la vida media de lamotrigina al doble. Se ha reportado también rash como parte de un síndrome de hipersensibilidad asociado con un patrón variable de síntomas sistémicos que incluyen fiebre, linfoadenopatía, edema facial y alteraciones sanguíneas y hepáticas. El síndrome muestra un amplio espectro de severidad clínica y puede en raros casos llevar a la coagulación intravascular diseminada (CID) y falla multiorgánica. Otros eventos adversos reportados como frecuentes (incidencia ≥ 1%) e infrecuentes (incidencia ≥ 1‰ y < 1%) durante todos los ensayos clínicos realizados en adultos y niños fueron: Generales: Frecuentes: dolor. Infrecuentes: daño accidental, reacciones alérgicas, dolor de espalda, escalofríos, edema facial, halitosis, infección y malestar. Aparato cardiovascular: Infrecuentes: enrojecimiento, tuforadas, hipotensión postural, taquicardia, vasodilatación, migraña, síncope y palpitaciones. Dermatológicas: Infrecuentes: acné, alopecia, piel seca, eritema, hirsutismo, rash maculopapular, decoloración de la piel, síndrome de Stevens-Johnson, sudoración, urticaria y rash vesiculolobuloso. Aparato digestivo: Infrecuentes: boca seca, disfagia, aumento de apetito, gingivitis, glositis, hiperplasia gingival, aumento de la salivación, alteraciones de los tests de función hepática, ulceraciones bucales, estomatitis, sed y trastornos odontológicos. Sistema hemolinfático: Infrecuentes: anemia, equimosis, leucocitosis, leucopenia, linfoadenopatía y petequias. Metabolismo: Infrecuentes: edema periférico, aumento de peso y pérdida de peso. Sistema musculoesquelético: Infrecuentes: trastornos articulares, miastenia y agitación. Sistema nervioso: Frecuentes: amnesia, confusión, hostilidad, disminución de la memoria, nerviosismo, nistagmo, alteraciones del pensamiento y vértigo. Infrecuentes: sueños anormales, agitación, trastornos de la marcha, akatisia, apatía, afasia, depresión del SNC, despersonalización, disartria, diskinesia, disforia, labilidad emocional, euforia, debilidad, estado de gran mal, alucinaciones, hiperkinesia, hipertonía, hipoestesia, aumento de la libido, taquipsiquia, espasmos musculares, mioclonus, ataques de pánico, reacciones paranoides, trastornos de la personalidad, psicosis, trastornos del sueño y estupor. Aparato respiratorio: Infrecuentes: disnea, epistaxis e hiperventilación. Sentidos especiales: Infrecuentes: trastornos de la acomodación, conjuntivitis, otalgia, fotofobia, trastornos del gusto y tinnitus. Aparato urogenital: Infrecuentes: galactorrea, hematuria, poliuria, incontinencia urinaria, aumento de la frecuencia urinaria, retención urinaria y moniliasis vaginal. PRECAUCIONES: Eventos dermatológicos: Se han comunicado casos graves de rash con hospitalización y discontinuación de lamotrigina. También se han comunicado algunos casos de muerte, pero son demasiado escasos como para poder estimar un porcentaje. El riesgo global de rash parece estar asociado con: Dosis inicial alta de lamotrigina (ver Posología). Exceso de los incrementos de dosis recomendados (ver Posología). Uso concomitante con valproato, el cual incrementa la vida media de la lamotrigina aproximadamente 2 veces. De todos modos, se han comunicado casos en ausencia de dichos factores. En ensayos clínicos, aproximadamente el 10% de los pacientes expuestos a la la motrigina desarrollaron rash. Estos cuadros asociados al uso de lamotrigina no parecen tener un patrón único de identificación. Típicamente, pueden aparecer entre la 2da. y la 8va. semana posterior al inicio del tratamiento. De todos modos, se han comunicado casos aislados luego de tratamientos prolongados (por ejemplo 6 meses). Por lo tanto, la duración de la terapia no debe tomarse en cuenta como un factor que permita predecir el riesgo potencial de aparición de rash. Si bien la mayoría de los cuadros de rash se resuelven luego de la suspensión del tratamiento, no es posible predecir cuáles de ellos pueden llegar a poner en compromiso la vida. Por lo tanto, la lamotrigina deberá suspenderse ante el primer signo de rash, a menos que la aparición del mismo claramente no esté relacionada con la administración del fármaco. La suspensión del tratamiento puede no prevenir las secuelas y/o el compromiso de la vida asociado al uso de lamotrigina. Muerte súbita inexplicable en epilepsia: Durante la evaluación de premarketing de la lamotrigina, se produjeron algunas muertes súbitas inexplicables entre la gran cantidad de pacientes que recibieron el medicamento. Algunos de estos casos pueden representar muertes asociadas con convulsiones en las cuales el episodio convulsivo no fue observable (por ejemplo durante la noche). La incidencia de estos eventos representa 0.0035 muertes por paciente - año. Si bien esta tasa excede la esperada en la población sana (considerando edad y sexo), se encuentra dentro del rango de la incidencia estimada de muerte súbita inexplicable en pacientes con epilepsia que no están recibiendo lamotrigina. Además, los valores de este tipo de eventos registrados en la población epiléptica que recibió otros fármacos antiepilépticos (aún químicamente no relacionados con la lamotrigina) fueron similares. Por lo tanto, estos datos parecen reflejar porcentajes de incidencia en la población más que un efecto de un fármaco en particular. Agregado de lamotrigina a un régimen multidosis con valproato: Reducción de dosis: dado que el ácido valproico reduce la depuración de la lamotrigina, la dosis de esta última en presencia de ácido valproico es menos de la mitad de la requerida. Uso en pacientes con enfermedad concomitante: Se aconseja precaución cuando se administra lamotrigina en pacientes con enfermedades que puedan afectar el metabolismo o la eliminación de la droga tales como: falla renal, hepática o cardíaca. En estudios de dosis únicas en sujetos con insuficiencia renal terminal, las concentraciones plasmáticas de lamotrigina no se encontraron significativamente alteradas. No obstante, dado que es posible una acumulación del metabolito glucurónido, se aconseja tener precaución cuando se indica el producto a pacientes con insuficiencia renal. Ligadura a tejidos oculares y otros que contengan melanina: Dado que la lamotrigina se liga a la melanina, puede acumularse en tejidos ricos en la misma. Esto hace posible que la lamotrigina pueda causar toxicidad en estos tejidos luego de su uso extendido. Pruebas oftalmológicas realizadas en estudios clínicos fueron inadecuadas para excluir daños después de la exposición prolongada. A pesar de que no hay recomendaciones específicas para el control oftalmológico periódico, debe tenerse en cuenta la posibilidad de efectos a largo plazo. Se recomienda monitoreo periódico de los parámetros renales, hepáticos y de coagulación, como también en pacientes con rash, dado que se han reportado casos asociados con neutropenia. Dosaje del fármaco: No ha sido establecida la utilidad del dosaje plasmático de la lamotrigina. Debido a que pueden producirse interacciones farmacocinéticas cuando se administra lamotrigina con otras DAEs, el monitoreo de ambos fármacos puede estar indicado, particularmente durante el ajuste de dosis. En general, el juicio clínico guiado por los valores plasmáticos de lamotrigina y otras DAEs deberá orientar el ajuste de dosis si fuera necesario. Carcinogénesis: No hubo evidencia de carcinogenicidad en estudios en ratas (2) y ratones (1), luego de la administración oral de lamotrigina hasta 2 años a la dosis máxima tolerada (30 mg/kg/día para ratones y 10 - 15 mg/kg/día para ratas, dosis equivalentes a 90 mg/m2 y 60 - 90 mg/m2, respectivamente). Las concentraciones plasmáticas fueron de 1 - 4 µg/ml en ratones y de 1 - 10 µg/ml en ratas. La concentración plasmática asociada con la dosis recomendada humana de 300 - 500 mg/día está en el rango de 2 - 5 µg/ml, pero se han registrado concentraciones tan altas como 19 µg/ml. Mutagenicidad: La lamotrigina no fue mutagénica en presencia o ausencia de activación metabólica cuando se probó en dos ensayos de mutación genética (la prueba de Ames y el ensayo in vitro del linfoma de ratones). En dos ensayos citogenéticos (ensayo linfocito humano in vitro y el de médula ósea en rata in vivo) la lamotrigina no aumentó la incidencia de anormalidades cromosómicas estructurales o numéricas. Fertilidad: No se detectó evidencia de efectos sobre la fertilidad en ratas que recibieron una dosis oral de lamotrigina hasta 2,4 veces la más alta dosis de mantenimiento humana de 8.33 mg/kg/día o 0.4 veces la dosis humana en mg/m2. No se conoce el efecto de la lamotrigina en la fertilidad humana. Embarazo: No se halló evidencia de teratogenicidad en ratas, ratones o conejos cuando se administró lamotrigina oral a animales preñados durante el período de organogénesis en dosis de 1.2; 0.5 y 1.1 veces en mg/m2, respectivamente, la dosis más alta de mantenimiento en humanos (es decir 500 mg/día). Sin embargo, se notó toxicidad materna y fetal secundaria produciendo reducción de peso y/o retraso en la osificación en ratones y ratas pero no en conejos con estas dosis. También se llevaron a cabo estudios teratogénicos con la administración intravenosa por bolo de la sal icetionato de lamotrigina en ratas y conejos. La administración intravenosa a ratas madres de una dosis de 0.6 veces la dosis más alta humana de mantenimiento, provocó un incremento de la incidencia de muerte intrauterina sin signos de teratogenicidad. Se realizó un estudio teratogénico de conducta en ratas medicadas durante la organogénesis. En el día 21 postparto las crías de hembras que recibieron 5 mg/kg/día o más, mostraron una prolongación significativa del período de latencia para la exploración a campo abierto y una menor frecuencia para pararse. En una prueba de laberinto en agua realizada en los días 39 a 44 postparto, las crías de las hembras que recibieron 25 mg/kg/día tardaron más tiempo para realizar la prueba. Estas dosis representan 0,1 y 0,5 veces la dosis clínica en mg/m2, respectivamente. La lamotrigina no afectó la fertilidad, la teratogénesis o el desarrollo postnatal cuando las ratas fueron medicadas antes y durante el apareamiento, y a través de la gestación y la lactancia con dosis equivalentes a 0,4 veces la dosis más alta de mantenimiento humano usual en mg/m2. Cuando se medicó a ratas preñadas por vía oral con dosis de 0.1; 0.14 ó 0.3 veces la dosis más alta de mantenimiento humano usual en mg/m2 durante la última parte de la gestación (días 15 - 20), se notó toxicidad materna y muerte fetal. En las hembras preñadas se redujo el consumo de alimentos y el incremento de peso, y se prolongó ligeramente el período de gestación (22.6 vs. 22.0 días en el grupo control). Se hallaron crías nacidas muertas en los 3 grupos tratados, con el mayor número en el grupo de altas dosis. También se notaron muertes postnatales, pero sólo en las dos dosis más altas y entre los días 1 - 20. Algunas de estas muertes parecen estar relacionadas con la droga y no ser secundarias a toxicidad materna. A pesar de que la lamotrigina no fue teratogénica en los estudios anteriores, disminuye la concentración fetal de folato en ratas, un efecto conocido asociado con teratogénesis en animales y humanos. No hay estudios adecuados y bien controlados en mujeres embarazadas. Ya que los estudios de reproducción en animales no siempre predicen la respuesta humana, la lamotrigina deberá ser usada en el embarazo sólo si el beneficio potencial justifica el riesgo potencial para el feto. Lactancia: Datos preliminares indican que la lamotrigina pasa a la leche humana. Dado que los efectos en el niño expuesto a la lamotrigina por este camino son desconocidos, este fármaco no debe utilizarse durante el amamantamiento. En caso estrictamente necesario se suspenderá la lactancia. Efecto sobre la capacidad de manejar y/o usar maquinarias: En estudios en voluntarios se ha demostrado que los efectos de la lamotrigina sobre la coordinación motora y visual fina, los movimientos oculares, los movimientos corporales y los efectos sedativos subjetivos no difirieron de los observados con placebo. No obstante, existen variaciones interindividuales en la respuesta a la terapia con DAEs y, en ensayos clínicos con lamotrigina, se han observado eventos adversos de tipo neurológico, tales como mareos y diplopía. INTERACCIONES: Con drogas antiepilépticas: El uso de estas drogas en combinación es complicado por la posibilidad de interacciones farmacocinéticas; la interacción de lamotrigina con fenitoína, carbamazepina y ácido valproico son reconocidas. Con excepción del ácido valproico, el agregado de lamotrigina a estas DAEs no afecta su concentración plasmática. El efecto de las combinaciones de DAEs en las concentraciones plasmáticas de cada una de ellas se resume en la siguiente tabla: Ver Tabla Efectos específicos de la lamotrigina en la farmacocinética de otras DAEs: Lamotrigina agregada a fenitoína: la lamotrigina no tiene efectos apreciables en la concentración plasmática de fenitoína. Lamotrigina agregada a carbamazepina: la lamotrigina no tiene efectos apreciables en la concentración plasmática de carbamazepina. Hay datos clínicos sugestivos de una mayor incidencia de mareos, diplopía, ataxia y visión borrosa en pacientes que toman carbamazepina con lamotrigina en relación a pacientes que reciben otras DAEs con lamotrigina. El mecanismo de esta interacción no está claro. En un pequeño grupo de pacientes (n=7) en un estudio controlado con placebo, la lamotrigina no tuvo efecto en la concentración plasmática de carbamazepina, pero en otro estudio no controlado pequeño (n=9), los niveles de carbamazepina aumentaron. Lamotrigina agregada a ácido valproico: cuando se administró lamotrigina a voluntarios sanos que recibían ácido valproico, la concentración en plasma en estado estable de DPA disminuyó un promedio de 25% sobre un período de 3 semanas y luego se estabilizó. Lamotrigina agregada a ácido valproico + fenitoína y/o carbamazepina: a pesar de que el efecto de la lamotrigina en los niveles plasmáticos de estas DAEs administradas en combinación no ha sido sistemáticamente evaluado, se espera que el efecto sea similar cuando se agrega lamotrigina a cada una por separado (DPA disminuye, PHT y CBZ sin cambios). Efectos específicos de otras DAEs en la farmacocinética de la lamotrigina cuando se adicionan a la misma: Fenitoína: el agregado de fenitoína disminuye la concentración de lamotrigina en 45 - 54% dependiendo de la dosis total diaria de fenitoína. Carbamazepina: el agregado de carbamazepina disminuye la concentración de lamotrigina en un 40%. Acido valproico: el agregado del mismo aumenta un poco más del doble la concentración de lamotrigina en voluntarios sanos. Fenobarbital o primidona: la adición de fenobarbital o primidona disminuye las concentraciones de lamotrigina en un 40%. Con anticonceptivos orales: en un estudio en voluntarias, la lamotrigina no afectó las concentraciones plasmáticas de etinilestradiol y levonorgestrel luego de la administración de las píldoras anticonceptivas. No obstante, como ocurre con la introducción de otras terapias crónicas en pacientes que toman anticonceptivos orales, debería ser evaluada cualquier modificación en el patrón del sangrado menstrual. Interacciones con otras drogas: Antagonistas del folato: la lamotrigina es un inhibidor de la dihidrofolato reductasa. Debe tenerse en cuenta esta acción cuando se indiquen otras medicaciones que inhiben el metabolismo del folato. Interacciones con pruebas de laboratorio: no se conocen. SOBREDOSIFICACION: Se han reportado casos de sobredosis de hasta 15 g de lamotrigina, algunos de los cuales resultaron fatales. Los casos de sobredosis presentaron síntomas de ataxia, nistagmo, aumento de convulsiones, disminución del nivel de conciencia, coma y retardo de conducción ventricular. No hay antídotos específicos para la lamotrigina. En caso de sospechar sobredosis, se recomienda la internación y la instrumentación de medidas generales de soporte, así como el monitoreo frecuente de signos vitales y observación cercana del paciente. Se puede inducir el vómito o proceder al lavado gástrico, pero debe recordarse que la lamotrigina se absorbe rápidamente. No es seguro que la hemodiálisis sea un medio eficaz para extraer la lamotrigina de la sangre. Ante la eventualidad de una sobredosificación, concurrir al hospital más cercano o comunicarse con los Centros de Toxicología: Hospital General de Niños "Dr. Ricardo Gutiérrez", Tel.: (011) 4962-6666/2247. Hospital General de Niños "Dr. Pedro de Elizalde", Tel.: (011) 4300-2115/ 4362-6063. Hospital Nacional "Prof. A. Posadas", Tel.: (011) 4654-6648/ 4658-7777. Hospital de Pediatría "Sor María Ludovica", Tel.: (0221) 451-5555. PROPIEDADES: El mecanismo exacto de la acción anticonvulsivante de la lamotrigina es desconocido. Un mecanismo propuesto involucra el efecto en los canales de sodio. Estudios farmacológicos in vitro sugieren que la lamotrigina inhibe los canales de sodio voltajes- sensitivos, estabilizando así las membranas neuronales con modulación consecuente de la liberación presináptica de aminoácidos excitatorios (por ej.: glutamato y aspartato). A pesar de que se desconoce su importancia en el uso humano, los siguientes datos caracterizan la actividad de la lamotrigina en ensayos de ligadura a receptores. La lamotrigina tiene un débil efecto inhibitorio en el receptor 5HT3 de serotonina (IC50 = 18 µM). No muestra una afinidad de ligadura alta (IC50 > 100 µM) a los siguientes receptores de neurotransmisores: adenosina A1, A2, adrenérgico α1, α 2 y ß, dopamina D1 y D2; gamma aminobutírico (GABA) A y B; histamina H1; opioide kappa; acetilcolina muscarínico y serotonina 5HT2. Los estudios no pudieron detectar un efecto de la lamotrigina en los canales de calcio dihidropiridina - sensibles. Tiene efectos débiles en el receptor opioide sigma (IC50 = 145 µM). La lamotrigina tampoco inhibió la captación de norepinefrina, dopamina, serotonina o ácido aspártico (IC > 100 µM). La lamotrigina in vitro inhibió la dihidrofolato reductasa, una enzima que cataliza la reducción de dihidro a tetrahidrofolato. La inhibición de esta enzima puede interferir con la biosíntesis de proteínas y ácidos nucleicos. Farmacocinética: La farmacocinética de la lamotrigina ha sido estudiada en pacientes con epilepsia: adultos, jóvenes, ancianos y voluntarios con insuficiencia renal crónica. Los parámetros farmacocinéticos se resumen en la siguiente tabla: Ver Tabla La depuración de la lamotrigina es afectada por la coadministración de drogas antiepilépticas. Es eliminada más rápidamente en pacientes que toman DAEs inductoras de enzimas hepáticas incluyendo carbamazepina, fenobarbital, fenitoína y primidona. Sin embargo, el ácido valproico disminuye la depuración de la lamotrigina (duplica el T1/2 de eliminación de la lamotrigina) administrado con o sin otras DAEs. Por lo tanto, la dosis de lamotrigina debe ser reducida a menos de la mitad cuando se administran juntas. Absorción: la lamotrigina se absorbe rápida y completamente luego de la administración oral con un metabolismo de primer paso casi inexistente (biodisponibilidad absoluta: 98%). La biodisponibilidad no es afectada por el alimento. La concentración plasmática pico ocurre de 1.4 a 4.8 horas luego de la administración. Distribución: el volumen de distribución luego de la administración oral va de 0,9 a 1,3 l/kg, es independiente de la dosis y es similar luego de la dosis única y/o múltiples en pacientes con epilepsia y en voluntarios sanos. Unión a proteínas plasmáticas: datos in vitro muestran que la lamotrigina está ligada a las proteínas plasmáticas en un 55% a concentraciones plasmáticas del fármaco de 1 a 10 µg/ml (10 µg/ml es 4 a 6 veces la concentración plasmática observada en los ensayos de eficacia controlados). Debido a que la lamotrigina no se une en gran medida a las proteínas plasmáticas, no es esperable que se produzcan interacciones significativas con otras drogas a este nivel. La unión de la lamotrigina a las proteínas plasmáticas no se altera en presencia de concentraciones terapéuticas de fenitoína, fenobarbital o ácido valproico. La lamotrigina no desplaza a otras drogas antiepilépticas (fenitoína, fenobarbital o carbamazepina) de sus sitios de unión a las proteínas plasmáticas. Metabolismo: la lamotrigina se metaboliza por conjugación con ácido glucurónico y su metabolito mayor es el conjugado 2-N-glucurónido que es inactivo. Luego de la administración oral de lamotrigina marcada a voluntarios sanos, se recuperó el 94% en orina y el 2% en heces. Las sustancias marcadas que se recuperaron en orina fueron lamotrigina inmodificada (10%), 2-N-glucurónido (76%), 5-N-glucurónido (10%), el metabolito 2-N-metil (0,14%) y otros metabolitos menores no identificables (4%). Inducción enzimática: los efectos de la lamotrigina sobre las familias específicas de enzimas oxidasas de función mixta no han sido sistemáticamente evaluados. Luego de administraciones múltiples (150 mg, 2 veces al día) en voluntarios normales que no estaban recibiendo otras medicaciones, la lamotrigina indujo su propio metabolismo, resultando en un descenso del 25% en la vida media y un aumento del 37% en la depuración plasmática en el estado estacionario en comparación con los valores obtenidos en los mismos voluntarios luego de una dosis única. Evidencia proveniente de otras fuentes sugiere que la autoinducción de la lamotrigina puede no producirse cuando este fármaco se administra como terapia coadyuvante en pacientes que están recibiendo otras DAEs inductoras de enzimas. Proporcionalidad de la dosis: en voluntarios sanos a los que se les administró dosis únicas y que no estaban recibiendo otras medicaciones en forma concomitante, las concentraciones plasmáticas de la lamotrigina aumentaron en forma directamente proporcional a la dosis administrada en el rango de dosis de 50 a 400 mg. En 2 estudios pequeños de pacientes con epilepsia que estaban recibiendo otras DAEs, también se registró una relación lineal entre las concentraciones plasmáticas de la lamotrigina en el estado estacionario luego de dosis de 50 a 350 mg, 2 veces al día. Eliminación: ver tabla 1. Poblaciones especiales: Pacientes con insuficiencia renal: en voluntarios con falla renal crónica (clearance de creatinina promedio 13 ml/min, rango = 6 a 23) y en individuos bajo hemodiálisis que recibieron una dosis de 100 mg de lamotrigina, las concentraciones plasmáticas promedio encontradas fueron de 42.9 horas (pacientes con falla renal crónica), 13 horas (durante la hemodiálisis) y 57.4 horas (entre hemodiálisis) en comparación con las 26.2 horas en voluntarios sanos. En promedio, aproximadamente el 20% (rango = 5.6 a 35.1) de la cantidad de lamotrigina presente en el organismo se eliminó por hemodiálisis durante una sesión de 4 horas. Pacientes con enfermedad hepática: la farmacocinética de la lamotrigina luego de la administración de una dosis única de 100 mg de lamotrigina se evaluó en 24 sujetos con disfunción hepática moderada a severa y se comparó con 12 sujetos sin deterioro hepático. La depuración aparente promedio de la lamotrigina fue de 0.31; 0.24 ó 0.10 ml/kg/min en pacientes con grado A, B, o C (clasificación de Child-Pugh de deterioro hepático), respectivamente en comparación con el valor de 0.34 ml/kg/min que se registró en los controles sanos. La vida media de la lamotrigina fue de 36, 60 ó 110 horas respectivamente en pacientes con grado A, B o C de deterioro hepático en comparación con el valor de 32 horas registrado en los controles sanos. Edad: Pacientes pediátricos: la farmacocinética de la lamotrigina luego de la administración de una dosis única de 2 mg/kg se evaluó en 2 estudios realizados en pacientes pediátricos con epilepsia. Todos los pacientes recibían en forma concomitante otras DAEs. Los parámetros farmacocinéticos de la lamotrigina en pacientes pediátricos se resumen en la tabla 2. Ver Tabla Ancianos: en un estudio de dosis única (150 mg de lamotrigina), la farmacocinética de la droga en voluntarios ancianos con edades comprendidas entre 65 y 76 años (clearance promedio de creatinina = 61 ml/min, rango = 33 a 108) fue similar a la farmacocinética de adultos jóvenes sanos en otros estudios. Sexo y raza: no se registraron diferencias en la depuración de la lamotrigina en función de sexo y/o raza. CONSERVACION: Condiciones de conservación y almacenamiento: Conservar en su envase original, en lugar seco, a temperaturas inferiores a los 30 °C. OBSERVACIONES: Mantenga este medicamento fuera del alcance de los niños. Este medicamento debe ser usado exclusivamente bajo prescripción y vigilancia médica y no puede repetirse sin nueva receta médica. Ante cualquier duda consultar al 0-800-444-2382 (BETA). Fecha de la última revisión: 07.02. PRESENTACIONES: Lagotran® 25 mg: Envase conteniendo 30 comprimidos dispersables. Lagotran® 50 mg: envase conteniendo 30 comprimidos dispersables. Lagotran® 100 mg: envase conteniendo 30 comprimidos dispersables. Lagotran® 200 mg: envase conteniendo 30 comprimidos dispersables. |
||
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |
||||
|
||||
 |
||||
|

|

|
| El contenido de este sitio está protegido por el derecho de propiedad intelectual, encontrándose prohibida su reproducción y distribución total o parcial por el medio que sea. | |